유전체 자료를 이용하여 실제 근교 계수 또는 혈연 계수를 구할 수 있다. 그러면 지금까지 혈통으로 구한 근교 계수 또는 혈연 계수는 실제가 아닌가? 그렇다. 혈통 정보에 기반하기 때문에 혈통이 없다면 또는 잘못되었다면 근교 계수 또는 혈연 계수가 0이 나오고, 혈통이 올바르다고 하여도 확률적인 근교 계수이다. 수 세대에 걸친 올바른 혈통이 있다면 확률적인 근교 계수와 실제 근교 계수의 차이가 크지 않을 것이다. 이 차이가 크다면 혈통이 올바르지 않거나 충분하지 않다고 생각할 수 있다. 여기서는 blupf90 family of programs의 하나인 pregsf90을 이용하여 유전체(실제) 근교 계수를 구해 본다. qcf90으로 구하면 좋겠지만 quality control과 근교 계수는 관계가 없는지 지금은 qcf90에서 구할 수 없다. pregsf90을 이용할 경우 renumf90 과정을 거쳐야 하고, 형식적이지만 자료 파일을 준비해야 한다는 귀찮음이 있다. 그래서 형식적인 자료로 단형질 개체 모형 예제의 자료를 이용한다.
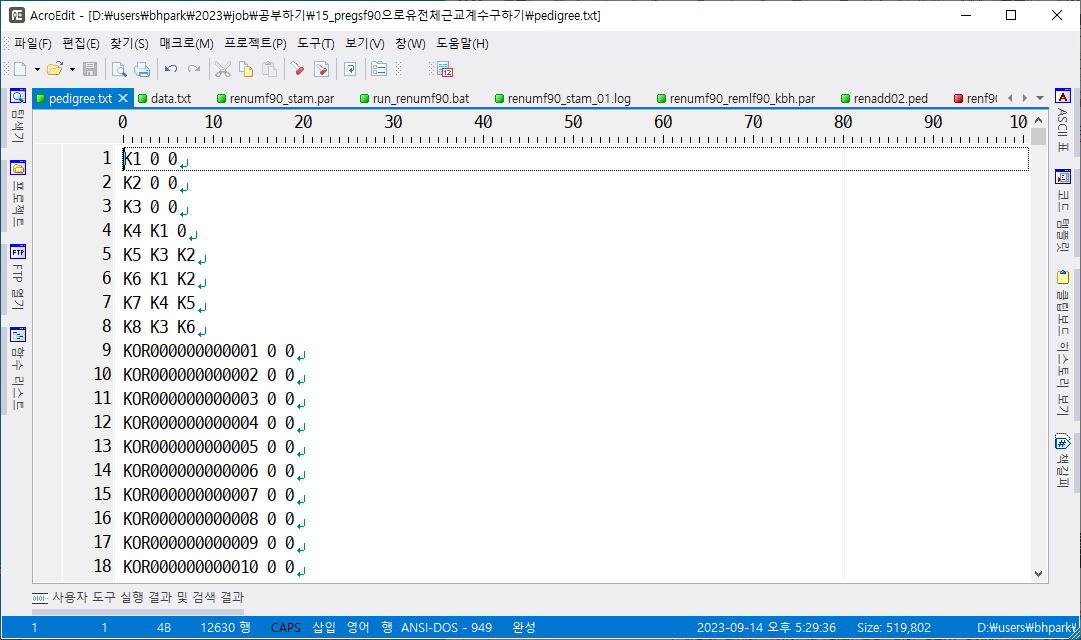
혈통(animal, sire, dam)
K1 0 0
K2 0 0
K3 0 0
K4 K1 0
K5 K3 K2
K6 K1 K2
K7 K4 K5
K8 K3 K6
위 혈통 자료를 우리가 구하고자 하는 혈통에 붙인다.
자료(animal, sex, gain)
K4 1 4.5
K5 2 2.9
K6 2 3.9
K7 1 3.5
K8 1 5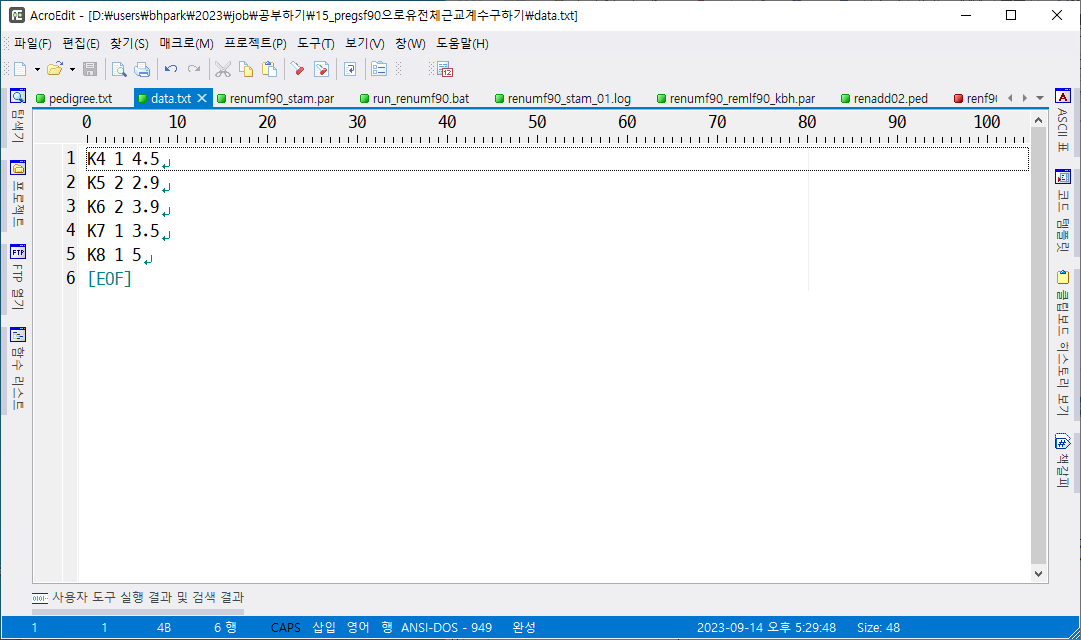
파라미터 파일
# Parameter file for program renf90; it is translated to parameter
# file for BLUPF90 family programs.
DATAFILE
data.txt
TRAITS
3
FIELDS_PASSED TO OUTPUT
WEIGHT(S)
RESIDUAL_VARIANCE
40
EFFECT
2 cross numer
EFFECT
1 cross alpha
RANDOM
animal
FILE
pedigree.txt
FILE_POS
1 2 3
SNP_FILE
kbh_hv1.txt
PED_DEPTH
0
(CO)VARIANCES
20
snp 파일도 포함되어 있고, PED_DEPTH를 0으로 하여 혈통 파일의 모든 개체를 분석에 포함한다.
실행
renumf90 renumf90_stam.par | tee renumf90_stam_01.log
실행을 하면 여러 파일이 생긴다.
renf90.par : 새로운 파라미터 파일
renf90.inb : 근교계수
renf90.dat : 리넘버된 data 파일
renf90.fields : renf90.dat에서 각 필드에 대한 설명
renadd02.ped : 리넘버된 혈통
kbh_hv1.txt_XrefID : 유전체 자료 ID의 리넘버. cross reference id 등과 같은 파일이 생긴다.
renf90.par 파일을 수정하여 renf90_pregsf90.par 로 저장한다.
# BLUPF90 parameter file created by RENUMF90
DATAFILE
renf90.dat
NUMBER_OF_TRAITS
1
NUMBER_OF_EFFECTS
2
OBSERVATION(S)
1
WEIGHT(S)
EFFECTS: POSITIONS_IN_DATAFILE NUMBER_OF_LEVELS TYPE_OF_EFFECT[EFFECT NESTED]
2 2 cross
3 12629 cross
RANDOM_RESIDUAL VALUES
40.000
RANDOM_GROUP
2
RANDOM_TYPE
add_an_upginb
FILE
renadd02.ped
(CO)VARIANCES
20.000
OPTION SNP_file kbh_hv1.txt
OPTION saveDiagHOrig
OPTION methodDiagH 1
OPTION saveGOrig
아래 세 줄을 추가한다.
“OPTION methodDiagH 1” 이 부분은 “OPTION methodDiagH 2”로도 분석할 수 있다.
SET OMP_STACKSIZE=64M
pregsf90 renf90_pregsf90.par | tee pregsf90_01.log

diagH.txt
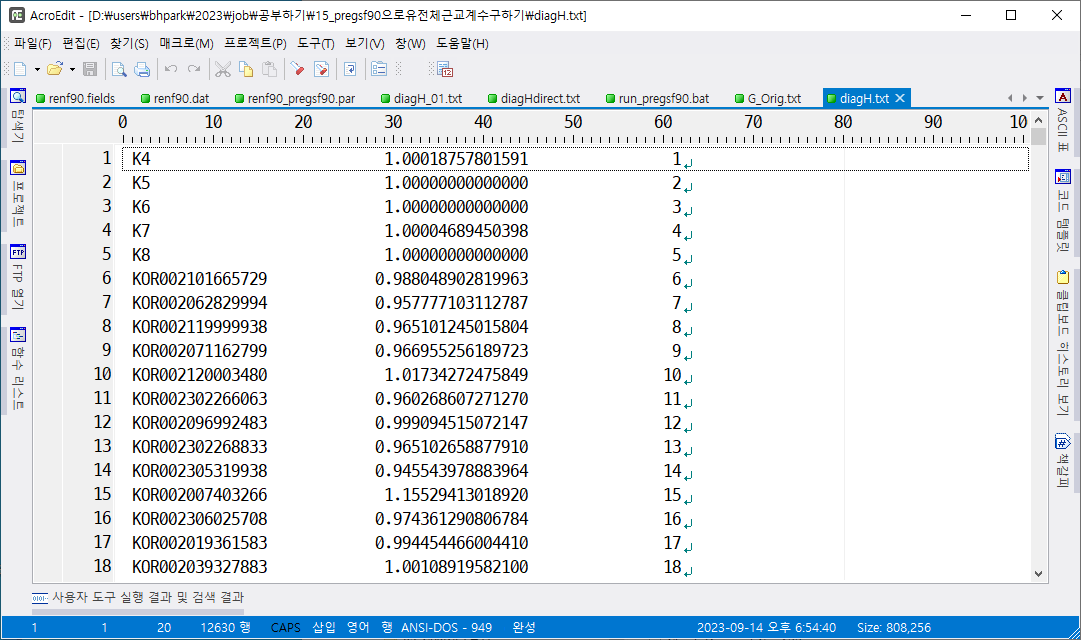
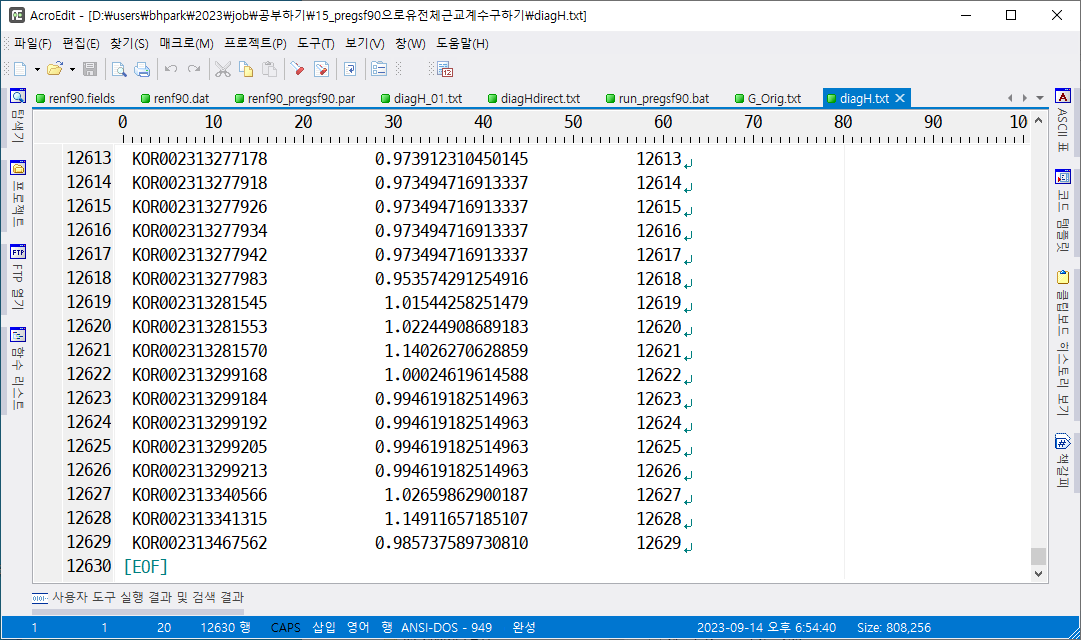
H matrix의 대각원소만 저장한 파일이다. 전 개체에 대해서 H matrix를 만들고 그 대각원소만 추출한 것이므로 이것들 중 유전체 자료를 가진 것만 추출하고 1을 빼면 유전체 근교 계수를 구할 수 있다.
OPTION methodDiagH 1을 OPTION methodDiagH 2로 변경하고 다시 실행해 보자.
실행
SET OMP_STACKSIZE=64M
pregsf90 renf90_pregsf90.par | tee pregsf90_01.log
diagHdirect.txt

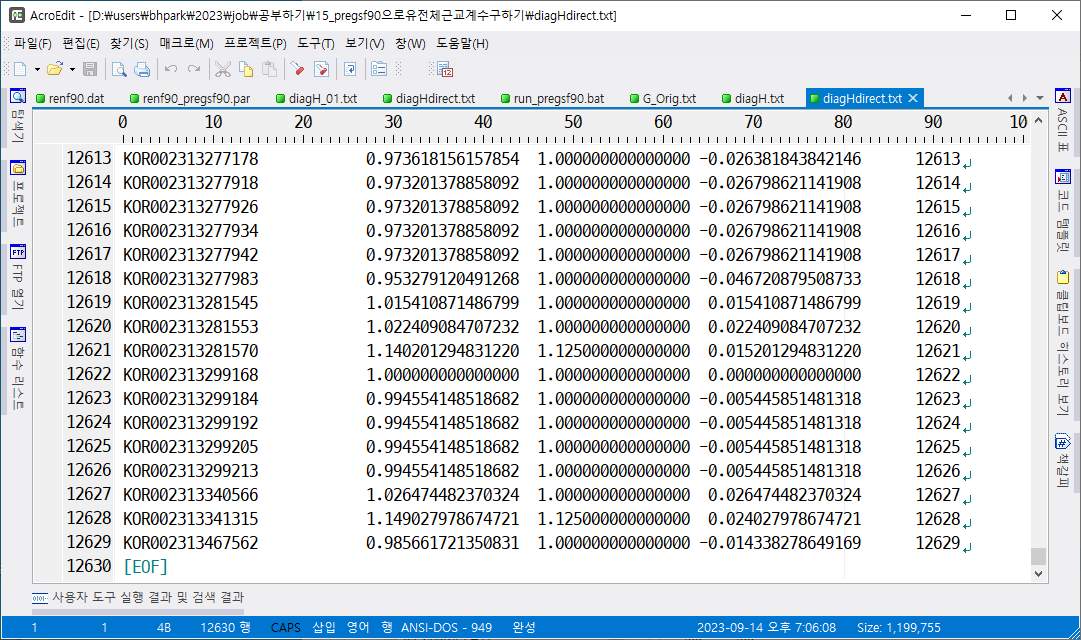
개체 ID, H matrix의 대각 원소, A matrix(혈통 기반 nemerator relationship matrix)의 대각 원소, 두 대각 원소의 차이.
A matrix의 대각 원소 값을 보면 혈통에 기반한 것이므로 1 또는 1.125등의 값을 보이지만, H matrix의 대각 원소는 유전체(실현) 근교 계수여서 매우 다양한 값을 볼 수 있다. 이 차이가 크다면 혈통 정보의 오류 또는 부족으로 볼 수 있다.
“OPTION methodDiagH 1”와 “OPTION methodDiagH 2”의 유전체 근교 계수를 비교해 보는 것도 좋을 것이다. 두 방법의 차이는 다음을 참조한다.
http://nce.ads.uga.edu/wiki/doku.php?id=readme.pregsf90
readme.pregsf90 [BLUPF90]
PreGSF90 / PostGSF90 PreGSF90 is an interface program to the genomic module to process the genomic information for the BLUPF90 family of programs This page also describes some options for PostGSF90 which is designed for genome-wide assocication study (GWAS
nce.ads.uga.edu
G_Orig.txt


G_Orig.txt 파일은 G matrix의 상삼각(또는 하삼각)을 보여준다. 이 값은 혈연 계수가 아니다. 혈연 계수는 근교 계수를 포함하여 계산한다. 하지만 근교 계수가 0이라고 가정한다면 혈연 계수라 볼 수 있다. 암튼 두 개체 사이의 관계 정도라 볼 수 있다. 혈통 정보가 부족할 경우 개체 사이의 혈연 관계 정도를 볼 수 있다.
유전체 자료는 있으나 혈통 정보가 부족한 소규모 집단의 계획 교배를 할 때 유전체 자료를 이용하여 개체의 근교 계수 또는 개체들 사이의 혈연 계수를 구하여 근친 교배를 피하고, 근교 퇴화를 예방할 수 있다.
'Animal Breeding > Genomic Selection' 카테고리의 다른 글
| 서로 다른 Chip 버전을 이용한 친자감정 및 부모 찾기 (1) | 2023.04.18 |
|---|---|
| H 행렬, H 행렬의 역행렬 유도 (0) | 2021.01.19 |
| SNP Effect를 이용한 DGV(or GEBV) 추정 (0) | 2020.03.05 |
| PostGSF90을 이용한 GWAS (0) | 2020.03.05 |
| SNP Marker Effect 추정 (0) | 2020.03.02 |