Quality Control(이하 QC)은 보유한 SNP 유전체 자료가 오류를 포함하고 있는지 아닌지 검사하는 것을 의미한다. BLUPF90 family program에서는 두 가지 프로그램을 제공하고 있다. pregsf90, qcf90. 두 프로그램이 하는 일을 거의 비슷한데 사용방법이나 목적에 약간의 차이가 있다. qcf90은 정말 유전체 자료 단독으로 또는 혈통을 포함한 유전체 자료에 대한 QC를 목적으로 한다. pregsf90은 유전능력평가 과정 중에 유전체 자료에 대해서 qc를 하고, 유전체 자료를 유전능력평가에 포함하는 역할을 한다. 그래서 qcf90 프로그램을 실행하기 위해서는 유전체 자료(혈통이 있으면 같이 분석 가능)만 있으면 된다. 그에 비해서 pregsf90은 표현형 자료, 혈통 자료, 유전체 자료를 가지고 renumf90 프로그램으로 renumbering후에 pregsf90을 실행할 수 있다. 여기서는 먼저 pregsf90을 이용하는 것을 살펴보자.
■ renumf90 실행
표현형 자료, 혈통 자료, 유전체 자료를 준비하고 renumf90을 실행해야 한다. 다음은 실행화면이다.

renumf_50k.par 의 내용
|
# BLUPF90 parameter file created by RENF90 DATAFILE data.dat TRAITS 5 6 7 8 9 FIELDS_PASSED TO OUTPUT
WEIGHT(S)
RESIDUAL_VARIANCE 517.52 0.72 0.33 0.18 0.13 0.72 708.47 0.50 0.36 0.08 0.33 0.50 34.54 0.19 0.11 0.18 0.36 0.19 6.72 0.19 0.13 0.08 0.11 0.19 1.43 EFFECT 1 1 1 1 1 cross alpha # animal RANDOM animal FILE pedi_03.txt SNP_FILE ..\04_50k_all\50k_for_pregs_01.txt PED_DEPTH 0 (CO)VARIANCES 221.79 0.65 0.12 0.25 -0.20 0.65 303.63 0.45 0.25 0.20 0.12 0.45 23.03 -0.20 0.30 0.25 0.25 -0.20 4.48 -0.02 -0.20 0.20 0.30 -0.02 1.43 EFFECT 2 0 0 0 0 cross alpha # fixed effect EFFECT 0 3 3 3 3 cross alpha # fixed effect EFFECT 0 4 4 4 4 cov # covariate OPTION chrinfo ..\04_50k_all\snp_map OPTION excludeCHR 30 31 32 OPTION saveCleanSNPs OPTION verify_parentage 1 OPTION callrateAnim 0.40 OPTION outcallrate OPTION outparent_progeny
|
옵션에 대한 자세한 설명은 매뉴얼을 참고하기 바란다. renumf90이 잘 실행되었으면 pregsf90을 실행할 수 있다.
■ pregsf90 실행
다음은 pregsf90 실행화면이다.

■ pregsf90.log에서 결과 보기
- SNP QC
|
Quality Control - SNPs with Call Rate < callrate ( 0.90) will removed: 811
Quality Control - SNPs with MAF < minfreq ( 0.05) will removed: 9873
Quality Control - Monomorphic SNPs will be removed: 1538 |
* call rate가 90% 미만인 SNP 811개 제거
* Minor Allele Frequency 가 5% 미만인 SNP 9873개 제거
* Monomorphic가 SNP 1538개 제거
- Animal QC
|
Quality Control - Removed Animals with Call rate < callrate ( 0.90): 0 |
* 개체의 call rate가 90% 미만인 개체가 있으면 삭제. 여기서는 없음
- 친자감정 결과 불일치
|
Checking Animals for Mendelian conflicts
Total number of effective SNP for checks on Animals: 37249
Number of Parent-Progeny Mendelian Conflicts: 246 Output stored in "Gen_conflicts" Output stored in "Gen_conflicts_all“
"Gen_conflicts" 과 "Gen_conflicts_all“를 잘 봐서 처리 |
- 유전자형 중복 개체
|
***************************************** * Possible genotype samples duplicates * *****************************************
i-j number of sample , i-j renumber Id, G(i,j), G(i,i), G(j,j), r(i,j) 213 21 14636 14487 0.9897 0.9905 0.9915 0.9987 29 22 14495 14488 0.3966 0.4322 0.4215 0.9292 57 22 14523 14488 0.3937 0.4287 0.4215 0.9261 57 29 14523 14495 0.3941 0.4287 0.4322 0.9156 99 98 14565 14564 1.0368 1.0397 1.0384 0.9978
3rd 컬럼과 4th 컬럼의 원래 id를 찾아서 유전자형 비교 가능성 1. 쌍둥이 2. 둘 중 하나의 유전자형이 틀림
확인 : 혈통, 생일 등을 확인하여 쌍둥이인지 확인하고, 쌍둥이가 아니면 이들이 다른 개체와 친자감정 일치가 났는지 검토, 쌍둥이라면 하나(다른 개체와 친자감정이 많이 안 된 개체)를 지우고, 한 개체만 다른 개체와 친자감정 일치가 나오면 그 개체는 놔 두고 다른 개체의 유전자형 삭제
|
- GRM의 대각성분 검토
|
Check for diagonal of genomic relationship matrix
** High Diagonal of genotype 218 1.68 Not Removed ** Low Diagonal of genotype 350 0.63 Not Removed ** High Diagonal of genotype 384 1.82 Not Removed ** High Diagonal of genotype 746 1.92 Not Removed ** High Diagonal of genotype 797 2.46 Not Removed ** High Diagonal of genotype 799 1.80 Not Removed ** High Diagonal of genotype 867 1.81 Not Removed
GRM의 대각성분의 값이 매우 크거나 매우 작은 경우. 잘못된 샘플(mislabeled sample), 다른 집단이나 계통에서 온 개체, 상태가 안 좋은 DNA 샘플로 실험하여 call rate가 매우 낮은데 이걸로 imputation해서 QC를 할 경우 등. 조언 : genome studio로 봤을 때 call rate가 90% 이하(또는 미만)인 개체는 아예 샘플 시트에서 빼서 읽어 오지도 않은 게 정신 건강상 좋음. |
- NRM과 GRM의 비대각 원소(off-diagonal elements)들의 상관
|
Off-Diagonal Using 50324 elements from A22 >= .02000
Estimating Regression Coefficients G = b0 11' + b1 A + e Regression coefficients b0 b1 = 0.008 0.904
Correlation Off-Diagonal elements G & A 0.845
A(NRM)에서 값이 0.02 이상인 비대각 원소만 뽑아서 GRM값과 상관을 구함. 이게 낮으면 혈통의 개체와 유전자형의 개체가 다르다고 볼 수 있음. 0.5 미만이면 경고 출력, 0.3미만이면 프로그램 중단. |
■ Gen_conflicts 파일 보기
|
Quality Control - Check Parent-Progeny Mendelian conflicts
Total animals: 54004 - Genotyped animals: 4319 - Effective: 4319
Number of pairs Individual - Sire: 3943 Number of pairs Individual - Dam: 0 Number of trios Individual - Sire - Dam: 0 Total number of effective SNP: 37249 Number of SNPs with Mendelian conflicts: 0 Total number of effective SNP for checks on Animals: 37249
Animal - Sire Conflict 396 520 2442 0.0480 Animal - Sire Conflict 573 1177 2829 0.0556 Animal - Sire Conflict 585 7658 2296 0.0451 Animal - Sire Conflict 586 2878 2520 0.0495 Animal - Sire Conflict 597 9566 2476 0.0486 Animal - Sire Conflict 604 7245 6727 0.1321
604 개체와 7245 아비가 친자감정 불일치가 났다는 얘기 604와 7245의 원래 번호를 찾아서 처리 방침 결정 -> 가능성 : 1) 604 또는 7245의 유전자형이 틀릴 수도 2) 604와 7245의 유전자형이 맞다면 혈통 정보가 틀릴 수도 확인 : 604 또는 7245가 다른 개체와 친자감정 일치가 났는지 조사,
다른 데서 친자감정 일치가 났는지 아닌지는 "Gen_conflicts_all“ 파일에서 확인
Number of Parent-Progeny Mendelian Conflicts: 2
#_Gen Renf90_Id #_sire tot_#_sire #_dam tot_#_dam #_ind tot_#_ind 336 341 2 10 0 0 0 0 1485 1490 0 0 0 0 1 2 1517 1522 0 0 0 0 1 2
* 2번째 부분 설명 - 1열 : SNP 파일에서 위치(행 번호) - 2열 : renumbered 된 개체 ID - 3열 : 부로서 conflict된 횟수 - 4열 : 부로서 비교된 횟수 - 5열 : 모로서 conflict된 횟수 - 6열 : 모로서 비교된 횟수 - 7열 : 개체로서 conflict된 횟수 - 8열 : 개체로서 비교된 횟수 |
■ Gen_conflicts_all 파일 보기
|
Animal - Sire 15 7491 13 0.0003 Match 일치 Animal - Sire 396 520 2442 0.0480 No_Match 불일치
각 개체가 몇 번 일치하는지 통계를 내어 유전자형이 올바른지에 활용
|
■ Principal Component Analysis 분석
- PCA 분석 실시를 위한 옵션
|
OPTION plotpca |
- pregsf90 로그 파일
|
Calculating PCA Eigenvalue Decomposition DSYEV LAPACK MKL #threads= 4 4 Elapsed omp_get_time: 1.5313 First 6 PC Eigenvalue % Explained PC: 1 29.75 1.862 PC: 2 25.96 1.625 PC: 3 20.63 1.291 PC: 4 18.84 1.179 PC: 5 17.12 1.071 PC: 6 16.89 1.057
* 각 PC의 값과 설명 범위 |
- 생성되는 파일 pc1vspc2
|
3.947475813628693E-002 2.685166110632073E-002 -4.991512155870371E-003 1.793240537447671E-002 -2.021474052015187E-002 -2.810828446416495E-002 -1.543435162734341E-002 -4.402118130908244E-002 -1.589047052620741E-002 -4.682080133059886E-002 ...
유전자형을 가지고 있는 개체들의 pc1과 pc2 출력 |
- f2
|
set title ' PC1 vs PC2' set nokey plot 'pc1vspc2' using 1:2 w points pt 6 ps .5 t ' pause 600 exit
gnuplot으로 그래프를 그릴 수 있게 해주는 스크립트 |
- gnuplot을 다운로드 받아 설치하고 실행

- 디레토리 변경 : 파일 -> change Directory

- pc1vspc2 파일을 이용하여 plotting


- f2 스크립트 파일로 그래프 그리기

- 쉘에서 f2 스크립트 파일로 그래프 그리기

- 분류에 따라 다른 색깔로 표시하기. 순서는 genotype 개체들의 순서와 같고 개체들을 분류하는 코드 입력. 여기서는 AB와 CD로 분류. geno_ani_group라고 저장

- 실행 옵션
|
OPTION extra_info_pca geno_ani_group 1 |
* 파일이름과 개체와 개체를 구별하는 컬럼이 1이라고 알려줌
- 결과 파일 1 : pc1vspc2t

* pc1과 pc2가 나오고 AB와 CD에 따라 구별하는 숫자가 3rd 컬럼
- 결과 파일 2 : f3, gnuplot을 이용하여 plotting을 하는 스크립트
|
set title ' PC1 vs PC2' plot 'pc1vspc2t' u 1:(($3==1 ) ? $2 : 1/0) w points pt 6 ps 1 t 'AB' , 'pc1vspc2t' u 1:(($3==2 ) ? $2 : 1/0) w points pt 6 ps 1 t 'CD' pause 600 exit
|
- gnuplot에서 f3 스크립트를 이용한 plotting
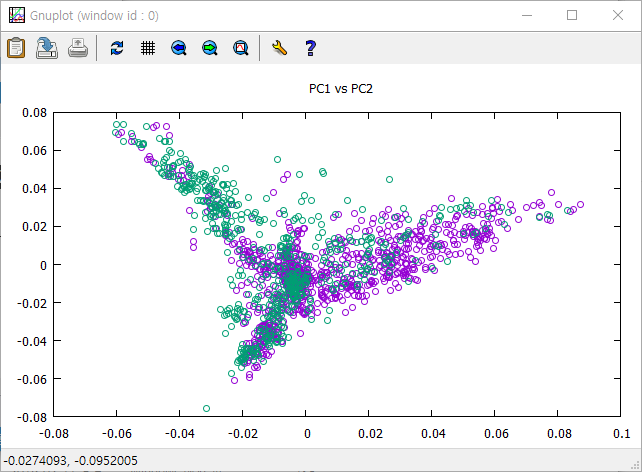
- 쉘에서 f3 스크립트를 이용한 plotting

■ OPTION saveCleanSNPs 옵션에 따라 생성된 파일
- 제거된 개체 : genotypefilename_Animals_removed
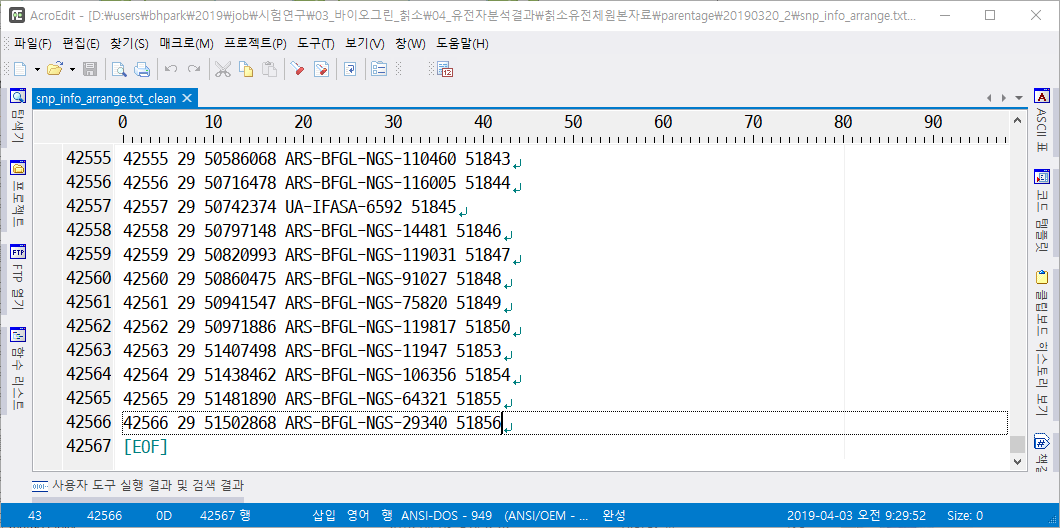
- QC된 SNP만 가지고 있는 파일 : genotypefilename_clean. 없어진 SNP가 있어 한 행의 끝이 42582임(=개체 15, 띄어쓰기 1, QC된 SNP 42566)

- QC된 후의 (즉 개체가 삭제된 후의) cross reference id 파일 : genotypefilename_clean_XrefID
- QC 때문에 제거된 SNP : genotypefilename_SNPs_removed, 9290개의 SNP가 제거됨

- QC 후의 SNP Map : snpmapfilename_clean, QC 후에 42566개의 SNP가 남아 있음
'Animal Breeding > Genomic Selection' 카테고리의 다른 글
| SNP 유전체 자료를 이용한 아비 어미 찾기 (0) | 2020.02.12 |
|---|---|
| QCF90이용한 quality control (0) | 2020.02.05 |
| FImpute를 이용한 SNP Imputation (1) | 2020.02.03 |
| illumina2pregs로 SNP genotype을 0, 1, 2로 변환하기 (0) | 2020.01.09 |
| GenomeStudio FinalReport 만들기 (1) | 2020.01.08 |